科技日报记者 刘霞
据美国麻 省理工学院(MIT)官网12日报道,该校科学家开发出一款名为EquiBind的几何深度学习模型,其将类药物分子与蛋白配对的效率比现有最快的计算分 子配对模型QuickVina2-W快1200倍。相关研究已经提交预印本服务器,并将提交给国际机器学习大会。
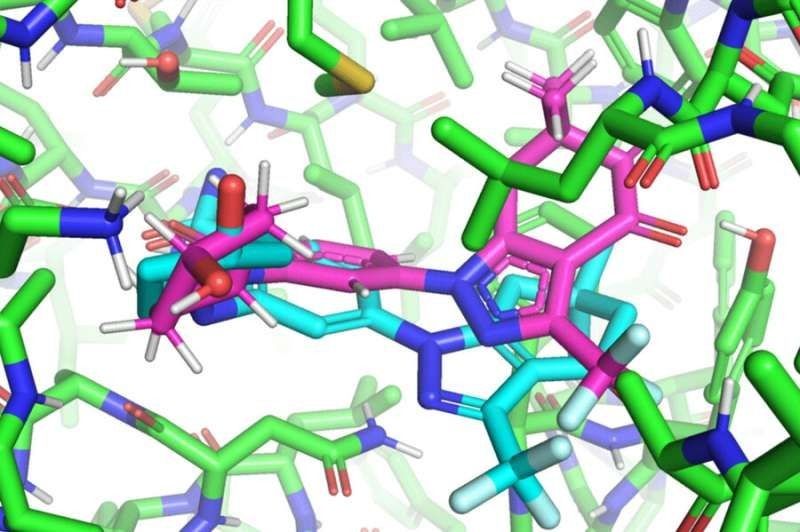
EquiBind(蓝绿色)能高效预测可与蛋白对接的配体。
图片来源:MIT网站
在药物开发之前,研究人员必须找到有潜力的类药物分子,这些分子可以与某些蛋白质靶点正确结合或“对接”——这一过程被称为药物发现。类药物分子(配体) 成功与蛋白质对接后,可以阻止蛋白质发挥功能。如果蛋白质是细菌的一种必需蛋白质,配体就可以杀死细菌,从而保护人体。
目前寻找潜在药物候选分子的计算过程大致如下:大多数最先进的计算模型依赖繁重的候选采样,以及评分、排序和微调等方法,从而让配体和蛋白质之间实现最佳“匹配”。
最新研究主要作者、MIT电气工程和计算机科学系研究生汉尼斯·斯塔克表示,上述传统的配体—蛋白质结合方法就像“尝试将钥匙插入有许多锁孔的锁中”。这 种方法需要花费大量时间对每个“锁孔”进行尝试,才能找到最佳匹配。相反,EquiBind仅需一个步骤就可以直接精准预测配体与蛋白质配对的精确位置, 这是因为其拥有内置的几何推理能力,可以帮助模型了解并学习分子的基本情况,在遇到新的数据时能够进行概括,以做出更好的预测。
该研究引起了专业人士的兴趣。接力医疗公司首席数据官帕特·沃尔特斯建议其团队在现有的一种用于肺癌、白血病和胃肠道肿瘤的药物和蛋白质上尝试这一最新模型,结果EquiBind取得了成功——而大多数传统的配对方法无法让蛋白和配体成功配对。
沃尔特斯说,EquiBind为配对问题提供了一种独特的解决方案,它结合了姿态估计和结合位点识别。这种方法利用了数千种公开可用的晶体结构的信息,可能对药物开发领域产生新的影响。